Page 10 of 17
SU24.2-3 | Pancreatic Endocrine Tumours — SDL Guide
Learning Objectives
- Classify pancreatic endocrine (neuroendocrine) tumours into functioning and non-functioning types and describe the major functioning syndromes — insulinoma, gastrinoma (Zollinger-Ellison) and the rarer entities (SU24.2).
- Describe the principles of investigation of pancreatic endocrine tumours — confirming the hormonal syndrome biochemically before localising the tumour — and the MEN-1 association (SU24.2, SU24.3).
- Outline the principles of management and prognosis of pancreatic endocrine tumours, integrating them into the overall disciplined approach to pancreatic disorders including pancreatitis (SU24.3).
INSTRUCTIONS
Pancreatic endocrine tumours are uncommon, often small, and easy to miss — yet they announce themselves through strikingly specific syndromes, because each arises from an islet cell and pours out the hormone that cell was built to make. A patient who repeatedly collapses with hypoglycaemia that resolves the instant they eat, or who has peptic ulcers that simply will not heal and unexplained diarrhoea, may be telling you, in the language of a single hormone, exactly which tumour they harbour. The discipline you have already learned for adrenal tumours and for pancreatitis applies here unchanged: confirm the hormonal syndrome biochemically first, localise the tumour second, and treat last. This module covers the functioning syndromes, their confirm-first work-up, the MEN-1 association to watch for, and the principles of surgical and medical management — completing the integrated picture of pancreatic disorders.
References
- Bailey & Love's Short Practice of Surgery, The Pancreas — Endocrine Tumours (textbook)
- SRB's Manual of Surgery, Pancreas — Endocrine Tumours of Pancreas (textbook)
- Sabiston Textbook of Surgery, The Pancreas — Neuroendocrine Tumors (textbook)
Version 2.0 | NMC CBUC 2024
CLINICAL SCENARIO
A 35-year-old woman is brought in confused and sweating for the fourth time in two months; each previous episode resolved within minutes of being given a sugary drink, and today her blood glucose is found to be dangerously low. Her relatives have started carrying biscuits everywhere. In another clinic, a 50-year-old man has been treated for peptic ulcers for two years, but his ulcers keep coming back, sit in unusual places, and are accompanied by stubborn watery diarrhoea that no one can explain. Neither patient has an obvious diagnosis on the surface, yet each is being driven by a tiny tumour in the pancreas that is broadcasting a single hormone — insulin in the first, gastrin in the second. The skill of endocrine pancreatic surgery is to hear the hormone in the story, prove it in the laboratory, and only then go looking for the small tumour responsible.
WHY THIS MATTERS
Pancreatic endocrine tumours matter out of all proportion to their rarity, for three reasons. First, they are often curable: a small benign insulinoma, once found, can be removed and the patient's life transformed. Second, they are dangerous when misread — recurrent hypoglycaemia can cause permanent neurological harm, and a gastrinoma's relentless acid can perforate the gut — so recognising the syndrome early genuinely saves patients. Third, they teach the same disciplined logic that governs all of endocrine and pancreatic surgery: the syndrome names the hormone, the hormone is confirmed biochemically, and only then is the tumour localised and treated. They also carry a vital warning: some are part of the inherited MEN-1 syndrome, so finding one should prompt a search for others. For a final-year student, the functioning pancreatic tumours are a satisfying example of bedside reasoning from physiology — each clinical picture is the predictable consequence of one islet hormone in excess.
RECALL
Recall the cells that make these tumours, because the tumour is simply that cell gone autonomous. The endocrine pancreas is organised into the islets of Langerhans, whose principal cell types are the beta cells (secreting insulin, which lowers blood glucose), the alpha cells (secreting glucagon, which raises it), the delta cells (secreting somatostatin, which inhibits other hormones), and cells producing other peptides. Recall too the integrated discipline you have already met for both adrenal tumours and pancreatitis: in any endocrine or pancreatic problem you confirm the hormonal or pathological diagnosis biochemically first, localise the lesion second, and intervene last — never operate on an image before the biochemistry is secure. Hold on to one organising idea: a functioning pancreatic endocrine tumour produces a clinical syndrome that is exactly the physiological action of its hormone taken to excess — so the syndrome itself points you straight to the cell of origin and to the test that will prove it.
The Patient with a Pancreatic Endocrine Tumour
Pancreatic endocrine tumours present in one of two fundamentally different ways, and recognising which one you are dealing with shapes the entire work-up. A functioning tumour declares itself through the syndrome of its hormone, often long before any mass is visible. The commonest functioning tumour is the insulinoma, which causes recurrent hypoglycaemia: episodes of confusion, sweating, palpitations, anxiety, altered behaviour or even seizures and loss of consciousness, characteristically occurring when fasting or exercising and relieved promptly by eating — the pattern captured by Whipple's triad (symptoms of hypoglycaemia, a documented low blood glucose at the time of symptoms, and relief of symptoms when glucose is given). The next classic functioning tumour is the gastrinoma, which causes the Zollinger-Ellison syndrome: severe, multiple, recurrent or atypically sited peptic ulcers that are resistant to standard treatment, together with diarrhoea (from the huge acid load) and reflux. Rarer functioning tumours produce their own signatures — a VIPoma causes profuse watery diarrhoea with hypokalaemia and dehydration (the WDHA syndrome), a glucagonoma causes a characteristic rash (necrolytic migratory erythema), weight loss and diabetes, and a somatostatinoma causes diabetes, gallstones and steatorrhoea. By contrast, a non-functioning tumour makes no clinically active hormone and therefore presents late, by mass effect (abdominal pain, obstructive jaundice) or by being found incidentally or as metastatic disease. The clinical story, in short, often tells you the hormone before any test is sent.
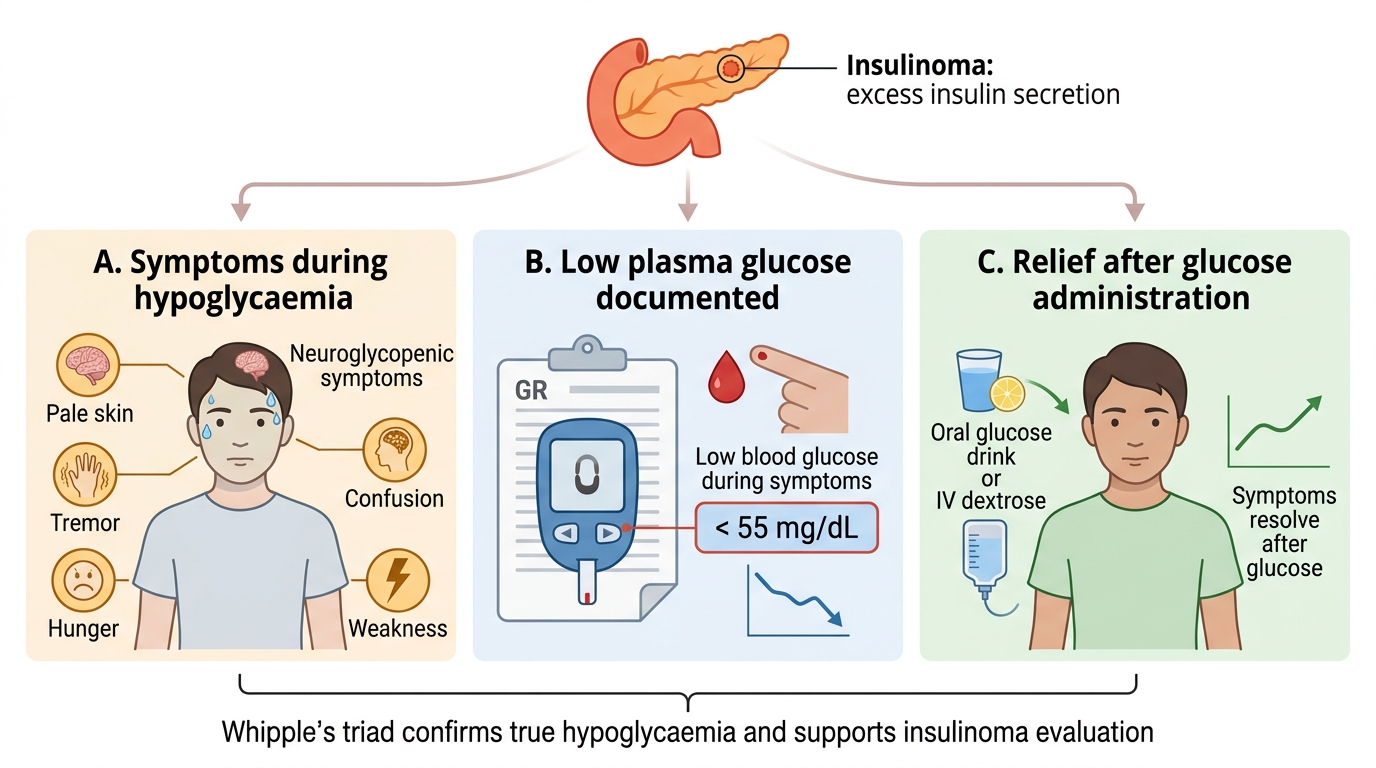
Whipple's Triad in Insulinoma
Classification and Pathological Basis
Pancreatic endocrine tumours — properly called pancreatic neuroendocrine tumours (PNETs) — arise from the islet cells of the pancreas, and they are classified first by whether they are functioning (secreting a hormone that causes a recognisable syndrome) or non-functioning (hormonally silent, presenting by mass or metastasis). Among the functioning tumours, the insulinoma is the commonest; it secretes insulin, causes fasting hypoglycaemia, and is usually benign (the great majority are solitary and non-malignant). The gastrinoma secretes gastrin to cause the Zollinger-Ellison syndrome, tends to occur in the so-called 'gastrinoma triangle' around the head of the pancreas and duodenum, and is more often malignant than the insulinoma, with a significant proportion having metastasised by diagnosis. The rarer functioning tumours each map to a hormone: the VIPoma (vasoactive intestinal peptide → watery diarrhoea, hypokalaemia, achlorhydria), the glucagonoma (glucagon → necrolytic migratory erythema, diabetes, weight loss), and the somatostatinoma (somatostatin → diabetes, gallstones, steatorrhoea); these are more often malignant. A crucial association to remember is the inherited multiple endocrine neoplasia type 1 (MEN-1) syndrome — the '3 Ps': Parathyroid hyperplasia (primary hyperparathyroidism), Pituitary adenoma, and Pancreatic (entero-pancreatic) endocrine tumours — so a pancreatic endocrine tumour, especially a gastrinoma or a tumour in a young patient, should prompt a search for the other components and a family history. The benign-versus-malignant tendency varies by tumour type, but the unifying principle is that each functioning PNET's syndrome is the direct, predictable excess of its islet hormone.
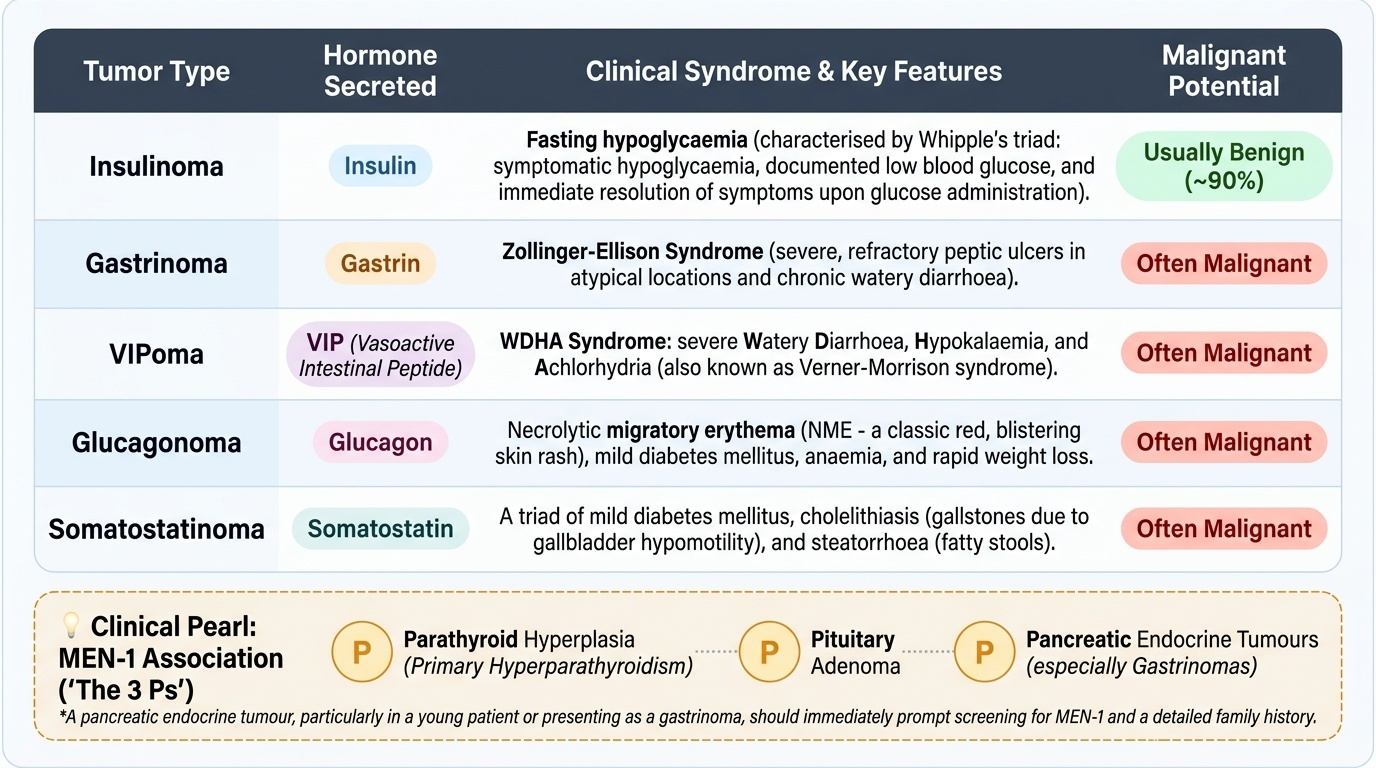
Provided image
| Tumour | Hormone | Syndrome | Malignant potential |
|---|---|---|---|
| Insulinoma | Insulin | Fasting hypoglycaemia (Whipple's triad) | Usually benign (~90%) |
| Gastrinoma | Gastrin | Zollinger-Ellison (refractory peptic ulcers + diarrhoea) | Often malignant |
| VIPoma | VIP | Watery diarrhoea, hypokalaemia, achlorhydria (WDHA) | Often malignant |
| Glucagonoma | Glucagon | Necrolytic migratory erythema, diabetes, weight loss | Often malignant |
| Somatostatinoma | Somatostatin | Diabetes, gallstones, steatorrhoea | Often malignant |
SELF-CHECK
A patient has recurrent fasting episodes of confusion and sweating, a documented low blood glucose during an episode, and prompt relief on eating. This is Whipple's triad. Which tumour and which confirmatory finding best fit?
A. Gastrinoma; raised fasting gastrin
B. Insulinoma; hypoglycaemia with inappropriately raised insulin AND C-peptide on a supervised fast
C. Glucagonoma; necrolytic migratory erythema
D. VIPoma; profuse watery diarrhoea
Reveal Answer
Answer: B. Insulinoma; hypoglycaemia with inappropriately raised insulin AND C-peptide on a supervised fast
Whipple's triad and fasting hypoglycaemia point to an insulinoma. It is confirmed by a supervised 72-hour fast showing hypoglycaemia with INAPPROPRIATELY elevated insulin and C-peptide. The raised C-peptide proves the insulin is endogenous (from the tumour), distinguishing it from surreptitious exogenous insulin administration.
Principles of Investigation
The investigation of a pancreatic endocrine tumour follows the same two-step discipline used throughout endocrine surgery: confirm the hormonal syndrome biochemically FIRST, and only then localise the tumour. For a suspected insulinoma, the diagnosis is confirmed by demonstrating Whipple's triad and then performing the gold-standard supervised 72-hour fast: the key finding is hypoglycaemia accompanied by inappropriately elevated insulin AND C-peptide, where the raised C-peptide is what proves the insulin is endogenous (made by the tumour) rather than surreptitiously injected, since manufactured insulin does not contain C-peptide. For a suspected gastrinoma, the diagnosis rests on a markedly raised fasting serum gastrin in the presence of high gastric acid output (a raised gastrin alone is non-specific because acid-suppressing drugs and achlorhydria also raise it, so the acid output matters). Only after the syndrome is biochemically secure does one localise the tumour, which is often small: the tools are cross-sectional imaging (CT or MRI), endoscopic ultrasound (EUS) — particularly good for small pancreatic lesions — and somatostatin-receptor imaging such as a gallium-68 DOTATATE PET or octreotide scan, which exploits the receptors these tumours express and is also useful for detecting metastases. Because of the inherited association, any patient with a pancreatic endocrine tumour should be screened for MEN-1 — checking serum calcium and parathyroid hormone for hyperparathyroidism and considering pituitary assessment and a family history. The same overarching framework applies to pancreatic disorders in general (SU24.3): a pancreatic problem is characterised biochemically and radiologically, its nature and extent defined, and treatment chosen on that secure basis rather than on an isolated scan finding.
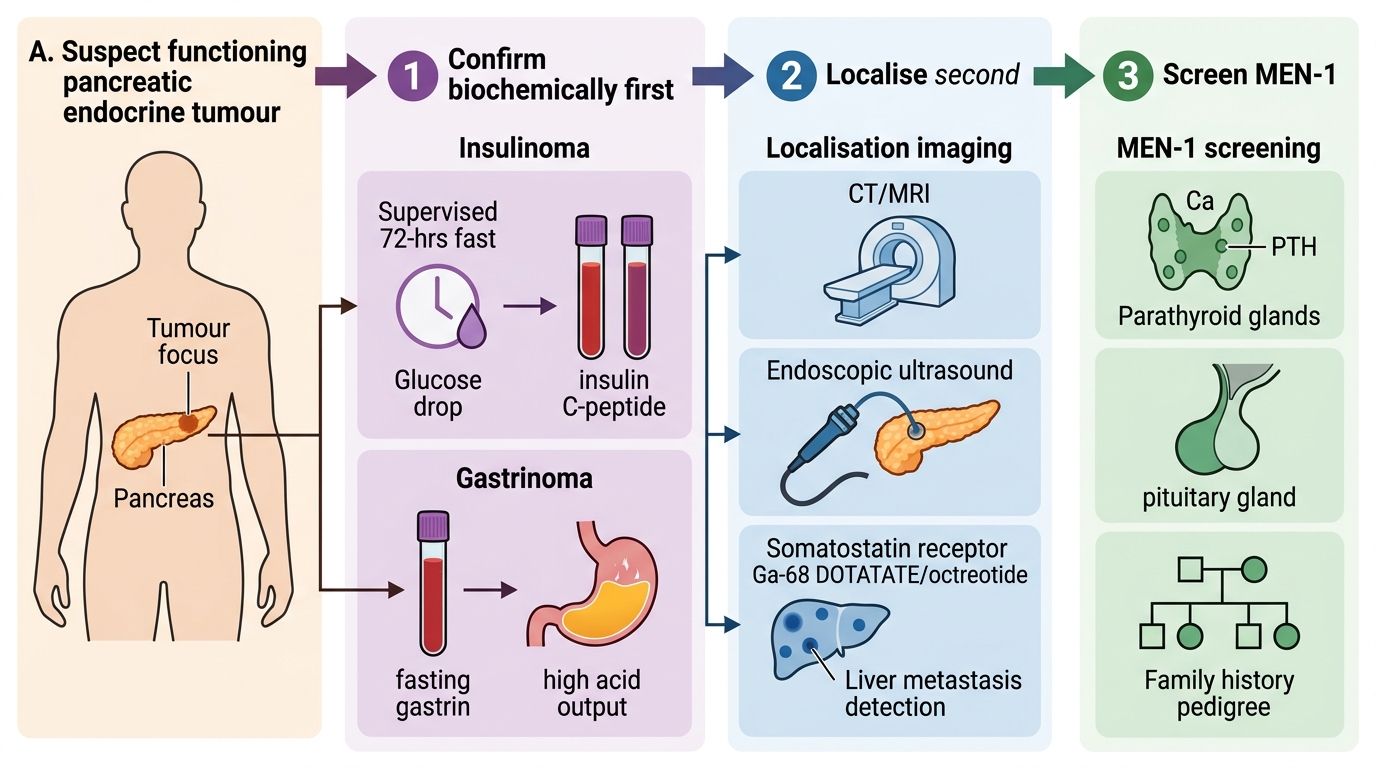
Functioning Pancreatic Endocrine Tumour: Confirm Then Localise
- Confirm first: insulinoma → 72-hour fast (hypoglycaemia + inappropriately raised insulin AND C-peptide); gastrinoma → raised fasting gastrin with high acid output.
- Localise second: CT/MRI, endoscopic ultrasound (small lesions), somatostatin-receptor imaging (Ga-68 DOTATATE/octreotide) — also detects metastases.
- Screen for MEN-1: calcium and PTH (parathyroid), pituitary assessment, family history.